您現在的位置是:首頁 >生活 > 2020-10-27 15:34:44 來源:
科學家打破分辨率記錄使用單粒子冷凍EM
查看蛋白質中原子的精確三維排列有助于我們了解其如何執行其功能。盡管近年來電子冷凍顯微鏡(cryo-EM)作為一種重要的結構生物學技術發展迅速,但X射線晶體學是唯一能夠可視化單個原子的技術。MRC分子生物學實驗室的Radu Aricescu和Sjors Scheres小組與Thermo Fisher Scientific和其他地方的科學家合作,現在已經能夠首次在三維低溫EM圖像中解析單個蛋白質原子。
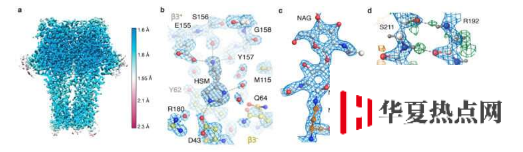
這項合作始于2019年初,當時Radu和Thermo Fisher Scientific的研究人員Abhay Kotecha希望在小膜蛋白樣品上測試新型cryo-EM硬件。選擇GABAA受體是Radu十年來的研究重點,因為使用最先進的技術可以達到的最高分辨率似乎已達到約2.5Ångströms(Å)的極限,但是顯然,更好的藥物設計需要更高的分辨率。
什么是原子分辨率?
分辨率通常以Ångströms報告,長度單位是十億分之一米或0.1納米,是指兩個物體之間可以看到的最小距離。
典型的碳-碳鍵的長度為1.5Å;蛋白質中的其他鍵短一些。因此,當分辨率降至1.2Å時,就有可能看到蛋白質中的單個原子,從而達到真正的原子分辨率。
在測試包括冷場發射槍電子源,新能量過濾器和新相機的新硬件開發時,該團隊還必須開發新的處理策略。Sjors小組的Jasenko Zivanov先前開發的光學像差校正算法,以及Chris Russo和Richard Henderson提出的算法,在從圖像中獲取最多信息方面起著關鍵作用。
Sjors小組的博士后Takanori Nakane收到了荷蘭埃因霍溫的Thermo Fisher Scientific公司的Abhay Kotecha在新顯微鏡硬件上收集的圖像后,在Radion和Andrija Sente以及Radu小組的其他成員中開發了最佳工作流程,此工作流程可處理GABAA受體圖像,同時反饋結果以快速優化顯微鏡設置。LMB科學計算團隊的Jake Grimmett和Toby Darling開發了一種新的大容量數據存儲系統,該系統提供了至關重要的支持來處理大約100 TB的數據。團隊的不懈努力導致了前所未有的1.7Å分辨率GABAA受體結構。
這是使用cryo-EM對除蛋白質載鐵蛋白以外的任何蛋白質樣品實現的最佳分辨率。載鐵蛋白通常被用作冷凍EM的基準,因為它的分子穩定性和24倍的對稱性允許從相對較少的顆粒進行高分辨率重建。
使用新的硬件和處理策略,該團隊能夠獲得1.22Å分辨率的脫鐵鐵蛋白結構,超過了以前的1.53Å記錄,是迄今為止獲得的最高分辨率的單粒子冰凍EM結構。最令人印象深刻的是,這種分辨率甚至可以使蛋白質結構內部的水分子上的單個氫原子可視化。蛋白質結構內部和藥物結合口袋中氫鍵網絡的可視化使研究人員可以更好地了解它們的工作原理。
這項工作代表了冷凍-EM作為結構生物學技術的關鍵障礙的突破,而新技術,數據收集和加工策略將擴大其結構可以解析為高分辨率的蛋白質的數量。這些更高分辨率的重建將使人們更好地了解蛋白質如何發揮作用,并有助于設計可能影響多種疾病治療的更具體的藥物。
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")